



- 端午肠胃 “减负” 正当时,菌小宝太空 1 号助力肠道轻盈畅爽
- 首届24节气养生文化节盛大启幕, 引领非遗非药物疗法新潮流
- 概念180内源修愈之旅:以“自愈力”重构抗老新叙事
- inne因你携手“大金条液体钙活力大使”刘晓庆,开启成人补钙新时代
精选导读




热门推荐
-
-

-
CLINIQUE LA PRAIRIE 瑞珀妮安吉抗衰理疗院尊享体验 匠心品质铸就 科学驱动健康
中国安吉,2024年5月28日:历经近一个世纪的辉煌历程和蓬勃发展,瑞珀妮在中国这片充满生机与活力的土地上继续展现其独特魅力..
2024-06-30
-
-
-
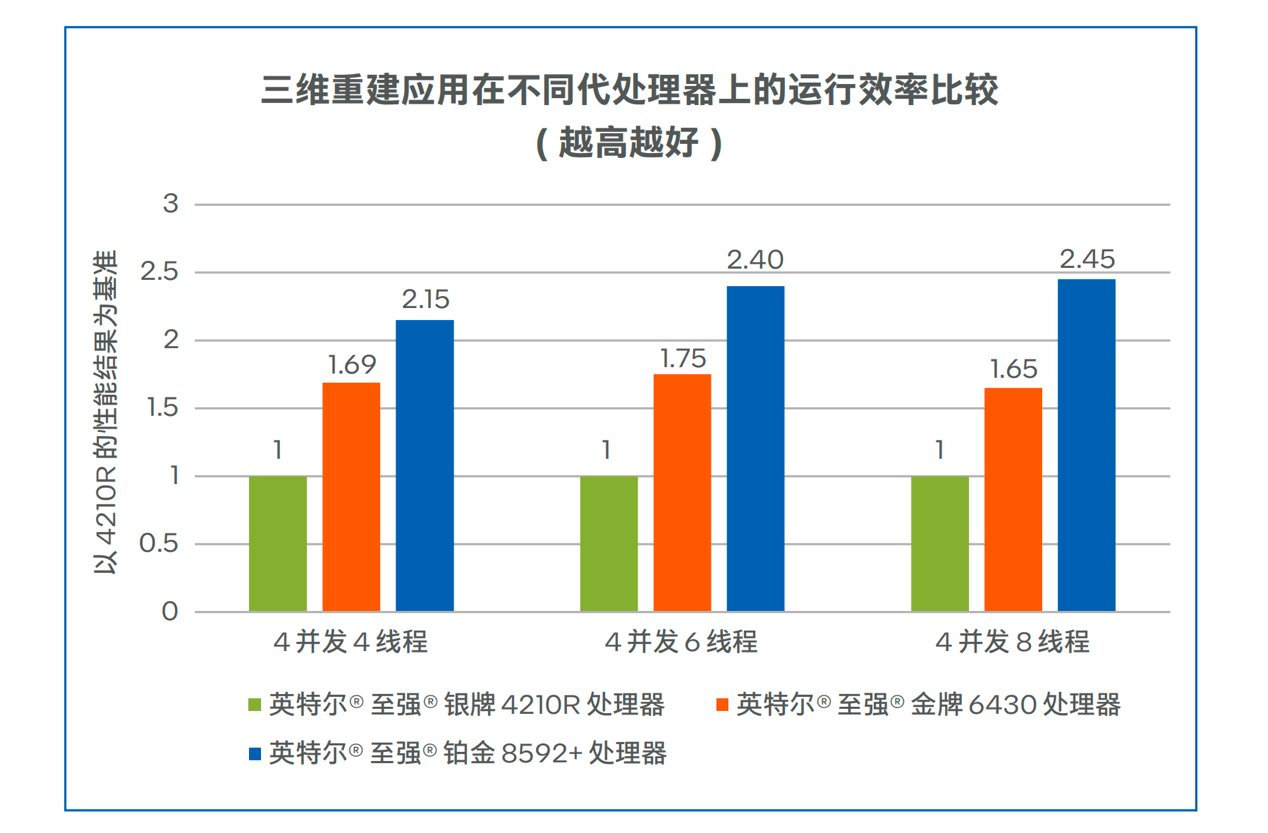
-
搞医学影像,为什么都青睐于CPU?
医学影像,越来越需要AI的帮助了。根据数据统计,目前我国医学影像数据年增速在30%,但影像科医生年增速却只有4%,医生面临较..
2024-05-31
-
-
-

-
CLINIQUE LA PRAIRIE 瑞珀妮安吉抗衰理疗院盛大开幕
中国安吉,2024年4月18日:备受瞩目的瑞珀妮安吉抗衰理疗院,在万众期待中迎来了隆重的开幕盛典。作为瑞士境外的首家抗衰理疗院,..
2024-04-26
-
-
-

-
汇聚党建力量·共筑健康堡垒公益健康论坛暨2024年全国肿瘤防治宣传周系列活动隆重举行
为认真落实党中央、国务院对癌症防治工作的决策部署,高质量推进健康中国建设,确保《健康中国行动癌症防治行动实施方案(20232..
2024-04-22
-
-
-

-
弘扬民族医药文化,助力健康中国 首届瑶医公益春晚在京隆重举行
乘龙聚势,绽放2024。为弘扬民族医药文化,助力健康中国建设,2024年2月2日,首届瑶医公益春晚在北京隆重举行,本次公益活动在..
2024-02-04
-
-
-

-
瑞珀妮宣布其在瑞士境外的第一家抗衰理疗院将于 2024年春在中国安吉焕新启幕
中国安吉,2023年12月25日:享誉国际的瑞士奢华长寿和医疗健康胜地--瑞珀妮(Clinique La Prairie,简称CLP)继续其全球布局,宣布与..
2024-01-30
-
-
-

-
北京瑶医医院院长覃迅云教授荣称“人民好医生”
2024年1月6日,第九届人民好医生典礼在京隆重举行。民建中央人口医药卫生委员会副主任、国际防癌长寿联盟主席、中国瑶医学科带..
2024-01-08
-
-
-

-
泰国BPK9国际医院:以高标准医疗服务,为来泰游客健康护航
自2023年1月8日中国政府允许中国公民出入境且免除入境隔离措施以来,出境游正在强势复苏。尤其是被列入首批允许团队出境游国家..
2023-05-26
-
头条阅读
端午肠胃 “减负” 正当时,菌小
- 在端午假期的餐桌上,糯米的黏糯、肉类的丰腴、甜食的甘美交织成一场味觉狂欢,然而狂欢过后还剩下很多的粽子还需要解决,但是........
首届24节气养生文化节盛大启幕,
- 5月28日,首届24节气养生文化节暨首届非遗非药物集市在太原湖滨会堂盛大开幕,现场气氛热烈非凡,吸引了众多市民和养生爱好者........
北京瑶医医院院长覃迅云教授当选
- 在医学发展的历史长河中,总有一些杰出的人物,以他们的智慧、勇气和不懈努力,为人类的健康事业开辟新的道路。民建中央人口医........
概念180内源修愈之旅:以“自愈
- 2025年5月9日,西班牙高端定制化内服抗衰品牌概念180(180 the concept)于西双版纳开启life coach内源修愈之旅。以和概念180........
inne因你携手“大金条液体钙活力
- 在健康中国战略实施的大背景下,国民对健康的关注度持续攀升,科学补钙已然成为现代健康生活方式的关键组成部分。顺应这一潮流........
热文榜单

最新快报